You must be signed in to read the rest of this article.
Registration on CDEWorld is free. You may also login to CDEWorld with your DentalAegis.com account.
Desquamative gingivitis (DG) is a group of clinical conditions characterized by intense redness (erythema) and gingival desquamation and ulceration. Clinical presentations range from mild cases with slight erythema and edema to severe cases involving blistering, erosion, and ulceration, often accompanied by pain and bleeding.1 Depending on severity, DG may be localized or widespread, including, in some cases, both intraoral and extraoral symptoms.1 Although DG does not directly cause periodontal attachment loss or alveolar bone destruction, it can significantly impair oral hygiene due to discomfort and bleeding, increasing the risk of long-term dental and periodontal issues.2 Additionally, systemic diseases associated with DG may result in both oral and extraoral symptoms, with some conditions leading to significant morbidity or even life-threatening complications.2 According to the 2017 American Academy of Periodontology and European Federation of Periodontology World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions, DG is classified as a non-plaque-induced gingival condition specifically linked to systemic diseases of immune origin.3
The most common causes of DG are oral lichen planus (OLP), mucous membrane pemphigoid (MMP), and pemphigus vulgaris (PV), which account for 75%, 9%, and 4% of cases, respectively.4 Other causes include autoimmune and allergic disorders like erythema multiforme (EM), graft-versus-host disease (GVHD), paraneoplastic pemphigus, systemic lupus erythematosus (SLE), and epidermolysis bullosa (EB).2 Contact irritants such as mouthwashes, toothpastes, dental materials, and medications may also trigger DG symptoms.1 Systemic treatments like corticosteroids or immunosuppressants in PV often improve gingival symptoms.2
Patients with DG often initially present with nonspecific symptoms, such as gingival bleeding, which may be misdiagnosed as plaque-induced gingivitis.2 Diagnostic delays are common, highlighting the need for early recognition of DG’s hallmark features. DG typically manifests between the fourth and sixth decades of life and most forms are common in females. Gingival lesions often appear early in diseases such as MMP and PV and sometimes may be the only clinical manifestation, particularly in MMP.2
Clinical Evaluation and Differential Diagnosis
While underlying conditions causing DG may have similar presentations, management differs based on the definitive diagnosis. Diagnoses of DG should involve a systematic approach that includes medical history review, clinical evaluation, laboratory tests, and histopathologic analysis.2
Medical history: When creating a differential diagnosis for DG lesions, it is critical to understand existing medical conditions and the onset and progression of lesions. Autoimmune disease diagnoses may indicate a predilection for other immune-modulated disorders as such conditions may cluster in patients.5,6 OLP and MMP tend to have a subacute onset that patients may not notice, whereas patients with PV can usually pinpoint when both intraoral and extraoral symptoms began.2
Patients should also be queried about events like infections and topical medicament use preceding DG symptoms. For instance, EM may be triggered by infection, while PV and SLE may be preceded by medication use. For localized DG lesions, a foreign body reaction should be considered, particularly if there is a history of trauma or dental procedures.2 Patient-reported symptoms, including standardized pain assessment, and the impact of medications on DG symptoms should also be assessed as they may inform DG management after diagnosis.
Intraoral clinical examination: Dental healthcare professionals should perform a thorough clinical examination to identify unique characteristics of various DG lesions to help aid in diagnosis. Practitioners may employ a cotton swab or air/water syringe to identify the Nikolsky sign—a clinical sign whereby pressure to the mucosa results in blisters and/or epithelial separation. Both MMP and PV commonly present with Nikolsky sign, though it is not often seen in OLP.2 Oral examinations should identify dental materials and their proximity to lesions, as some restorative materials (eg, amalgam, composites, cobalt, gold) can induce oral lichenoid reactions. Such reactions may mimic OLP but generally resolve after the removal of the material. If suspected, skin patch testing can confirm hypersensitivity.
Assessment of extraoral involvement: Evaluating extraoral involvement in DG-associated disorders is crucial for interprofessional communications and management. If patients have not been previously diagnosed, they should also be screened for autoimmune diseases and inflammatory bowel diseases.5,6
Clinical Presentations for Underlying Diseases
Many forms of DG affect oral and extraoral sites, including the skin, scalp, nails, and distant mucosal tissues. Gingival manifestations generally present as erythema, erosions, and ulcerations. While intact vesicles or bullae may occur, they tend to rupture quickly in the mouth. For most forms of DG, definitive diagnosis requires histopathologic and immunopathologic studies. However, characteristic oral or skin lesions may help practitioners formulate differential diagnoses.2
A summary of clinical and histologic findings is provided in Table 1.
Oral Lichen Planus
OLP is a chronic mucocutaneous disorder affecting 1% to 2% of middle-aged adults, with up to a 3:1 female:male presentation. OLP typically presents as bilateral and symmetric lesions. While it is sometimes classified as an autoimmune condition, its exact cause remains unknown. OLP is hypothesized to result from abnormal activation of CD4+ T-lymphocytes, leading to its characteristic clinical features.7 Common forms of OLP include8:
Reticular/keratotic: White striations or papules, usually painless, on the buccal mucosa, tongue, lips, and gingiva. Post-inflammatory hypermelanosis may occur in darker-skinned individuals.
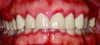
Erythematous/erosive/atrophic: Painful, red, raw mucosa, often on the gingiva (desquamative gingivitis), coexisting with reticulated areas (Figure 1).
Ulcerative: The most painful form, with yellow fibrin membranes.
Bullous: Rapidly rupturing bullae leading to erosive lesions from mucosal trauma.
Skin involvement occurs in 10% to 15% of cases and may include nail dystrophy. Genital–gingival lichen planus is often associated with desquamative OLP, with buccal mucosa fibrosis seen in a minority of patients.8
Mucous Membrane Pemphigoid
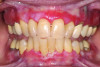
MMP is a chronic, immune-mediated disorder affecting mucous membranes.9 It predominantly affects middle-aged females, commonly presenting as painful, erythematous, ulcerated gingiva.8 The oral mucosa, especially the gingiva, is the most frequently affected site, although ocular, nasal, pharyngeal, and genital mucosa may also be involved (Figure 2). Symptoms range from mild lesions to severe systemic disease.9 Oral signs include bullae that rupture into ulcers, DG, and mucosal scarring.9 In addition to oral MMP presentation, ocular symptoms are also frequently observed. Ocular MMP may present with chronic conjunctivitis, burning, irritation, photophobia, and excessive tearing.9 In extensive MMP cases, esophageal or laryngeal involvement and subsequent scarring may cause stenosis, strictures, or airway obstruction.2
Pemphigus Vulgaris
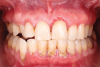
PV is a severe autoimmune disease caused by autoantibodies against desmosomes, leading to intraepithelial clefting and the formation of vesicles and bullae on the skin and mucosa (Figure 3). In approximately 70% of PV cases, the condition may be triggered by medications or vaccinations. PV mainly affects middle-aged adults, particularly those of Mediterranean, Ashkenazi Jewish, or South Asian descent, with a prevalence of about 1 in 5 million individuals. Serum autoantibodies are typically present in patients with skin involvement but less common in those with only oral manifestations.8 If untreated, PV can be fatal.2
Other Conditions Associated With Desquamative Gingivitis
Less common conditions include EM, GVHD, paraneoplastic pemphigus, SLE, and EB. EM is an acute skin condition often triggered by infections or medications, with target-shaped lesions and possible mucosal involvement. GVHD occurs after stem cell or bone marrow transplants, where transplanted immune cells attack the recipient’s tissues. Paraneoplastic pemphigus is a rare autoimmune disorder linked to malignancies, causing mucosal erosions and skin blisters. EB is a genetic condition causing fragile skin that blisters easily, with severity ranging from mild to widespread lesions and scarring.8 Medication-induced SLE occurs when certain drugs, such as hydralazine, procainamide, and isoniazid, trigger an autoimmune response, leading to lupus-like symptoms that typically resolve after discontinuing the offending medication.2
Pathogenesis and Histologic and Immunologic Findings
In addition to histopathologic evaluation, immunofluorescence studies are essential for detecting pathologic immunoreactants in DG-associated disorders. These studies can be performed using tissue samples via direct immunofluorescence (DIF) or serum via indirect immunofluorescence (IIF). Immunoreactants typically include immunoglobulins (IgG, IgM, IgA), complement factors, and fibrinogen, with correlations to specific autoantigens involved in each disease. Immunofluorescence is crucial to establish a definitive diagnosis as typical features may not be visible under light microscopy.
Oral lichen planus: OLP is characterized by a dense, linear band of lymphocytes beneath the surface epithelium, often leading to the migration of lymphocytes from the connective tissue into the epithelium, causing basal layer degeneration.2 This indicates T-cell-mediated etiology, particularly involving cytotoxic CD8+ T cells, which induce apoptosis in oral epithelial cells through tumor necrosis factor (TNF)-alpha release.4 CD4+ T cells also contribute by producing inflammatory cytokines. This process results in a lymphocytic infiltrate, basal cell liquefaction, and either hyperkeratosis or thinning of the epithelium, which helps confirm the diagnosis.2
Key histologic features also include basal cell degeneration or squamatization, a lymphocytic band at the interface, and possible thickening or blurring of the basement membrane zone.2 Subepithelial clefting from basal cell loss does not reflect clinical blister formation. OLP lesions may be parakeratotic or hyperkeratotic with acanthotic, atrophic, or eroded epithelium. Other features include spongiosis, lymphocyte transmigration, colloid body formation, sawtooth rete ridges, and a lymphocytic band at the interface.2
DIF is useful for diagnosing OLP and shows “shaggy” fibrinogen deposition at the basement membrane, with IgM labeling of colloid bodies. IIF is typically negative.
Mucous membrane pemphigoid: MMP is characterized by subepithelial separation caused by autoantibodies targeting hemidesmosomes that anchor the epithelium to the connective tissue.4 Diagnosis relies on clinical presentation, histopathology, and DIF. Biopsy, including lesional and intact tissue, is necessary for diagnostic histology, as ulcerated areas may show nonspecific inflammation. Hematoxylin and eosin (H&E) staining typically reveals a subepithelial cleft with eosinophils, neutrophils, and lymphocytes infiltrating the lamina propria.2
Microscopically, MMP shows subepithelial clefting with basal cells protruding into the cleft. The epithelium may be thickened or thinned with minimal spongiosis. Biopsies often show detached epithelial strips with an intact basal layer and minimal inflammation. Direct immunofluorescence reveals linear IgG, C3, and/or IgA at the basement membrane, with immunoreactants localizing to the lesion roof in salt split-skin tests.8 Indirect immunofluorescence detects circulating antibodies in 44% of mild cases and 100% of severe cases, with IgA often associated with more severe, treatment-resistant disease.8
Pemphigus vulgaris: PV is characterized by intraepithelial separation caused by autoantibodies targeting desmosomal proteins, particularly desmogleins (Dsg3 in mucosal lesions and Dsg1 in cutaneous lesions). Histologic features of PV include acantholysis, ie, the separation of keratinocytes in the spinous layer, Tzanck cells, and a “tombstone” appearance in the basal layer.4 Diagnosis is based on clinical presentation, histopathology, immunopathology, and serology. Histopathology shows suprabasal cleavage with acantholysis, while DIF reveals intercellular IgG and C3 deposition, which is the gold standard for diagnosis.2 Serological tests, including IIF and enzyme-linked immunosorbent assays (ELISA), detect circulating autoantibodies. PV diagnosis requires the presence of IgG deposition on epithelial cell surfaces by DIF,3 along with clinical criteria such as flaccid blisters and intraepidermal blisters with acantholysis. IIF and ELISA can detect anti-Dsg3 and/or anti-Dsg1 antibodies, helping guide diagnosis and treatment.3,8
Clinical Treatment Recommendations
Patients with DG frequently report mucosal sloughing, gingival bleeding, and oral discomfort, especially when consuming acidic or spicy foods and beverages. Topical steroids, with or without initial systemic steroid therapy, are the primary treatment for rapid disease control. A soft plastic stent that covers the gingiva is helpful for holding the steroid under occlusion, although gauze can also be used for this purpose.10 Studies have shown improvement when patients adhere to strict oral hygiene protocols and receive regular professional cleanings, with the best results achieved when combined with topical corticosteroid therapy.10
Oral lichen planus: OLP treatment is generally based on symptom severity and mucosal involvement. No treatment is recommended for asymptomatic OLP. Topical corticosteroids (rinses, gels, or sprays) are a first-line treatment for mild OLP gingival and erosive lesions, with rinses preferred due to limited systemic impacts.9 If the OLP is persistent, higher-potency topical anti-inflammatory formulations or systemic therapies (corticosteroids, calcineurin inhibitors, or retinoids) may be considered. Nonsteroidal treatments such as tacrolimus and apremilast may be used for managing chronic ulcers. Caution should be exercised, however, as tacrolimus carries a black box warning regarding its potential carcinogenic risk. Additionally, the recommendations supporting its use may be outdated, and newer treatment options with improved safety profiles should be considered. For patients with skin/genital lesions, seeking care with dermatology and/or rheumatologic specialists and hepatitis C screening are recommended. Regular follow-ups are essential, and patients should be informed of a slight risk (0.1% to 1%) of oral squamous cell carcinoma transformation of OLP lesions.8,10
Mucous membrane pemphigoid: Extraoral MMP involvement (conjunctiva, larynx, genital areas) raises concerns about scarring.2 Treatment focuses on symptom control, scar prevention, and maintaining function using topical, intralesional, and systemic therapies.2 Dermatology and ophthalmology referrals are recommended for screening and management of MMP cases.10 First-line treatments include topical corticosteroids or calcineurin inhibitors.2 Severe cases may require systemic therapies like corticosteroids, dapsone, and rituximab. Further, anti-infectives like dapsone and minocycline may be effective for mild and/or oral lesions. Monitoring for medication side effects, including immunosuppression, is crucial.
Pemphigus vulgaris: PV lesions can range from small vesicles to large bullae, leading to ulceration and pain. Intraorally, the soft palate is most frequently affected, followed by the buccal mucosa, tongue, and lower labial mucosa.
Systemic corticosteroids remain a primary PV treatment, but long-term monitoring of overall exposure and dosage is critical to minimize side effects. PV remains chronic with no cure, and therapy focuses on symptom relief and mucosal healing. Topical corticosteroids are first-line therapy for oral lesions, with calcineurin inhibitors as second-line treatments. Referral to a dermatologist is recommended, even without skin lesions, as oral symptoms can precede cutaneous involvement by up to a year. “Steroid-sparing” strategies, combining corticosteroids with immunosuppressive agents like azathioprine, cyclophosphamide, or methotrexate, help reduce long-term steroid use. Monoclonal antibodies and/or immunosuppressants may be considered for long-term control.9
Topical antifungal medications may prevent iatrogenic candidiasis, common with intraoral steroid use. Patients should maintain long-term follow-up to monitor oral health and PV progression and response.10
Conclusion
DG is a nonspecific clinical manifestation linked to oral and systemic conditions and allergic reactions to local factors making diagnosis and treatment challenging. Management includes avoiding irritants, maintaining oral hygiene, and using topical or systemic corticosteroids for severe cases. Proper evaluation through history, examination, biopsy, and histology, along with timely referrals for extraoral evaluations, is essential.
Dental healthcare professionals may identify mucocutaneous lesions initially, but the nonspecific nature of oral lesions can lead to misdiagnosis. Improved knowledge of immune-mediated oral diseases enhances diagnosis and patient care. Early recognition, specialist referrals, and interdisciplinary communication are key for timely intervention.
Collaboration between dental clinicians and medical specialists is crucial for managing conditions like OLP, MMP, and PV, which require coordinated care. Dentists can focus on early detection and local management of oral presentations, while medical specialists can provide systemic therapy and monitoring. A multidisciplinary approach ensures optimal patient outcomes.
ACKNOWLEDGMENT
The clinical photographs (Figure 1 through Figure 3) were provided courtesy of Lauren Miele Dasher, DMD, MS.
ABOUT THE AUTHORS
Priscilla Sosa, DMD
Periodontal Resident, University of Alabama at Birmingham School of Dentistry, Birmingham, Alabama
Maria L. Geisinger, DDS, MS
Professor, Acting Chair, Director, Advanced Education Program in Periodontology, Department of Periodontology, University of Alabama at Birmingham School of Dentistry, Birmingham, Alabama; Diplomate, American Board of Periodontology
Queries to the author regarding this course may be submitted to authorqueries@conexiant.com.
REFERENCES
1. Maderal AD, Salisbury PL 3rd, Jorizzo JL. Desquamative gingivitis: clinical findings and diseases. J Am Acad Dermatol. 2018;78(5):839-848.
2. Lo Russo L, Fedele S, Guiglia R, et al. Diagnostic pathways and clinical significance of desquamative gingivitis. J Periodontol. 2008;79(1):4-24.
3. Holmstrup P, Plemons J, Meyle J. Non–plaque-induced gingival diseases. J Clin Periodontol. 2018;45 suppl 20:S28-S43.
4. Shaqman M, Hamdan A, Karadsheh O, et al. Desquamative gingivitis: a challenging diagnosis for clinicians. Br Dent J. 2020;229(1):26-30.
5. Antonelli E, Bassotti G, Tramontana M, et al. Dermatological manifestations in inflammatory bowel diseases. J Clin Med. 2021;10(2):364.
6. Nylund K, Helenius-Hietala J, Åberg F, et al. Persistent oral mucosal lesions preceding diagnosis of Crohn’s disease and primary sclerosing cholangitis. Clin Case Rep. 2023;11(5):e07226.
7. Lavanya N, Jayanthi P, Rao UK, Ranganathan K. Oral lichen planus: an update on pathogenesis and treatment. J Oral Maxillofac Pathol. 2011;15(2):127-132.
8. Woo SB. Granulomatous, immune mediated, and autoimmune conditions. In: Oral Pathology. 3rd ed. Philadelphia, PA: Elsevier; 2023:202-225.
9. Patel S, Kumar S, Laudenbach JM, Teruel A. Mucocutaneous diseases: oral lichen planus, mucous membrane pemphigoid and pemphigus vulgaris. J Calif Dent Assoc. 2016;44(9):561-570.
10. Peters SM. Demystifying desquamative gingivitis: diagnosis and management. Decisions in Dentistry. 2020;6(6):40-43.